{kind=link}
About Us
We're Fast
PSI4 uses the latest techniques like density-fitting, and is optimized for multi-core execution of DFT, MP2, SAPT, and coupled-cluster.
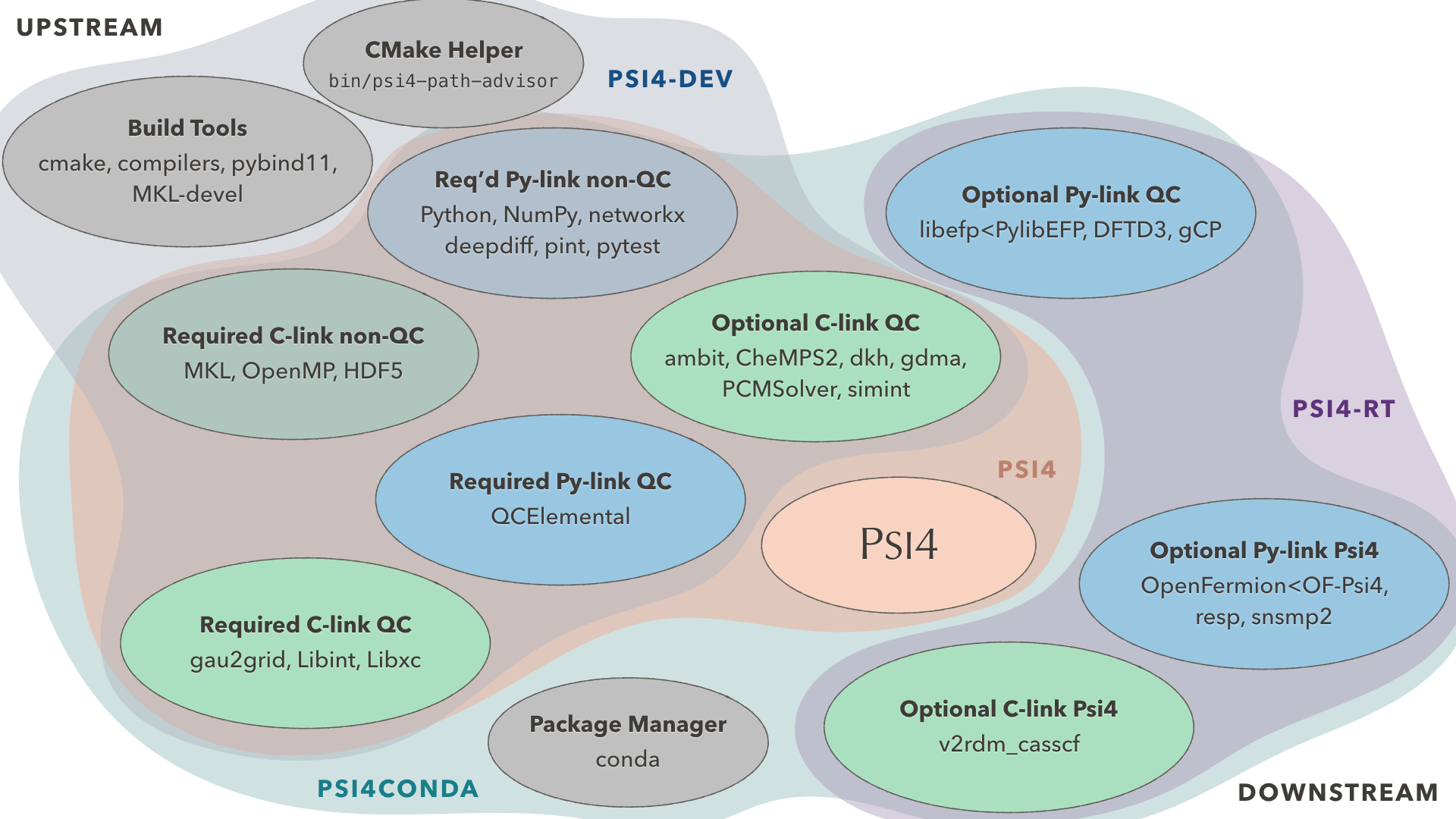
We're Modular
PSI4 is a C++/Python core that easily interfaces with and is extended by standalone community projects in our growing Software Ecosystem.
{kind=link}
We're Open Source
PSI4 is free and distributed under the truly open-source LGPL3 license. Use in education, research, and industry is encouraged.
We're Easy to Use
PSI4 uses simple input files and automates common procedures like basis set extrapolation and counterpoise correction.
We're Robust
PSI4 checks code changes with continuous integration and code coverage tools, and automatically keeps documentation up-to-date.
We're Developer-Friendly
PSI4 can be loaded as a Python module, so you can integrate it into your workflow. Plugins make it easy to extend features.
Get Started with Psi4
With computationally demanding portions in modern C++, exports of many classes into Python via Pybind11, and a flexible Python driver, PSI4 strives to be friendly to both users and developers.
Downloads Tutorials Programming Psi4EducationRecent Updates








After installing a new psi4conda with psi4 1.3rc2, threading issue is now solved. I can often get close to 1600% CPU usage and walltime of job is substantially reduced [from v1.1].
The SAPT code is lightning fast and simple to use. The input file structure is also clean, simple, and intuitive.